What is Rett Syndrome?
Imagine the symptoms of Autism, Cerebral Palsy, Parkinson’s, Epilepsy, and Anxiety Disorder all in one child.
Rett Syndrome is caused by a random mutation in a gene called MECP2. When born, babies appear to be normal but then, typically between 6 and 18 months, symptoms begin to appear. Many children with Rett Syndrome are unable to speak or walk, develop breathing and heart rhythm abnormalities, suffer from seizures, tremors, anxiety, gastro-intestinal issues, and orthopedic difficulties such as severe scoliosis, hip dislocations, and ankle/foot positioning problems.
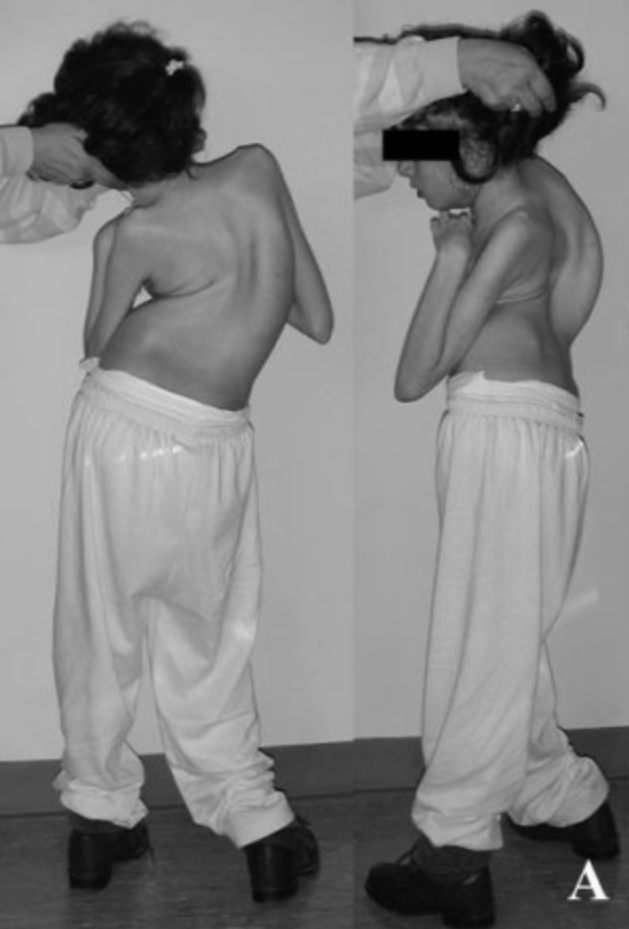
Image of severe scoliosis in rett syndrome
The hallmark symptom of Rett Syndrome is the loss of purposeful hand use, replaced by near constant hand-wringing, clapping, and mouthing.
Rett Syndrome primarily occurs in girls. Boys with Rett Syndrome tend to have more severe symptoms unless another genetic difference is present that actually gives them a better chance for survival.
Medical research has discovered that, unlike other neurological disorders, Rett Syndrome is not degenerative. In animal models, there is a dramatic reversal of symptoms when the defective MECP2 gene is corrected (see this video of a mouse before reversal and after reversal). Human clinical trials are expected to begin in 2019 pending FDA approval. There is an expectation that the reversal of symptoms seen in the animal models will also be seen in humans.
The advancements in technology have also led to a change in the previous understanding that children with Rett Syndrome will not advance intellectually past that of a 9 month old. While it is possible that there is some intellectual disability, it appears to not be nearly as devastating as previously thought.
Rett Syndrome does not cause a reduction in life span but because it is so physically debilitating, other diseases can present more of a risk to life than happens in the “typical” population.
People suffering from Rett Syndrome are trapped in a body that just doesn’t work for them.
Rett Syndrome History
In 1954, Dr. Andreas Rett observed two young girls in the waiting room of his office making similar hand washing motions with their hands. Upon further study, he saw that their developmental and medical histories were also remarkably similar. He soon began searching for other children with the same non-typical behavior and histories, traveling throughout Europe to find them.
Unbeknownst to Dr. Rett, Dr. Bengt Hagberg in Sweden noticed the same thing in some of his young female patients in 1960.
Despite Dr. Rett publishing his findings in German and English language medical journals in the 1960’s and 1970’s, very few people took notice of this growing body of evidence of a newly identified disorder. It wasn’t until 1983 when Dr. Hagberg published his report in the Annals of Neurology, did it finally catch the interest of medical researchers. In honor of Dr. Rett, this disorder was named Rett syndrome.
Rett Syndrome Research Breakthroughs
A team of researchers at Baylor University in Houston, Texas and a team at Stanford University in Palo Alto, California focused their efforts on trying to find the cause of Rett Syndrome. Finally, in 1999, Ruthie Amir, a researcher at Baylor University lab headed by Dr. Huda Zoghbi, discovered that a mutated gene on the X chromosome was the cause of a majority of cases of Rett syndrome. Identifying this disorder to be an X-linked disorder helps explain why it seemed like Dr. Rett and Dr. Hagberg only found young girls during their search for similarly affected children. This major breakthrough provided a critical clue allowing researchers to now focus on a specific gene, a clue many other disorders don’t have. Learn more about Dr. Huda Zoghbi here.
Armed with the knowledge that the gene, known as MeCP2, was responsible for Rett syndrome, Dr. Adrian Bird at the University of Edinburgh, created the first mouse model that would allow researchers to study Rett syndrome and begin the task of understand what role the MeCP2 gene played which could then lead to treatments and a cure. There have been subsequent improvements to the mouse model, which were initially all male, including the ability to create female mouse models.
In 2007, Dr. Bird discovered that the symptoms of Rett syndrome found in the mouse models reversed when the MeCP2 gene was returned to normal, even in adult mice. This provided a proof-of-principle that the reversal of Rett syndrome symptoms may be possible in humans as well.
But there were significant challenges to proving this same principle in humans. To correct the MeCP2 gene in humans required that the gene be repaired in the cells in the brain. The brain protects itself with special types of cells that surround the brain, preventing most molecules from entering. Only very small molecules and some gases can freely pass. The only other way would be to surgically get past the barrier which is very risky and would most likely not provide more than localized results where the surgical site is.
The discovery of the gene and the subsequent research has revealed ways to address the symptoms using treatments focused on those symptoms. In 2012, a phase 1 trial using a growth hormone known as IGF-1 was started to try to treat some specific Rett syndrome symptoms, but also to have a positive impact generally for all Rett syndrome symptoms by helping to increase synaptic activity and reduce inflammation in the brain. The phase 1 and phase 2 trials have been completed, and enrollment for the phase 3 trials is going on now. There appears to be some significant positive results and some encouraging anecdotal reports from families that it does have a positive impact.
In 2014, AveXis, a Novartis Company, discovered a virus that was able to cross the blood brain barrier allowing it to be potentially used for gene therapy where entering the nervous system was critical. In 2018, they reported on the phase 1 trial results for Spinal Muscular Atrophy with remarkable results. A problem still existed though because the complete MeCP2 gene was too large for this virus to carry. Further research has resulted in a better understanding on what parts of the MeCP2 gene are important in order to reduce the size of the material that needs to be included in gene therapy treatments.
What Does Crush Rett Syndrome Do With the Funds It Raises?
Our mission is to raise awareness and funds to crush Rett Syndrome out of existence.
2016
- Charity established in March.
- A few fundraising events were completed to raise funds, awareness, and nurture supporters.
- Networked with other Rett Syndrome charities, examined the current research projects, and committed to financially support some starting in 2017.
2017
- A few fundraising events were completed.
- The goal to double the 2016 revenue was achieved.
- $60,000 sent to Rett Syndrome Research Trust directed towards specific work on a gene therapy approach to cure Rett Syndrome.
- Examined current gene therapy research as well as research to re-awaken the inactive X chromosome to allow the healthy MECP2 gene to counteract the effects of the defective gene.
2018
- A few fundraising events were completed.
- Plans for a larger fundraising event have begun for 2019.
- 2 additional board members added to the Board of Directors.
- $75,000 sent to Rett Syndrome Research Trust directed towards specific work on genetic, RNA, and other approaches to cure Rett Syndrome.
2019
- A larger fundraising event is in the implementation phase and will launch in Q3, 2019.
- Examination of the various promising curative research efforts is being done with the expectation that direct funding of one or more of these research projects will be done in 2019.